Glycogen storage disease (GSD) is a rare genetic disorder that affects about one in 20,000 people in the U.S.[*]. People with GSD have trouble synthesizing and breaking down glucose, which can cause a laundry list of health issues, including chronic low blood sugar, enlarged liver, weak muscles, and more.
And although glycogen storage disease is inherited, it’s not necessarily a hopeless diagnosis. Gene therapies show early promise, and enzyme replacement drugs appear to help some of the GSD population with enzyme deficiency. Nutrition changes may also offer some relief. Read on to find out.
What Is Glycogen Storage Disease?
Glycogen storage disease is a metabolic disease that affects your ability to synthesize or break down and use glycogen — the storage form of glucose (more on this in the next section).
With a few exceptions, most forms of GSD inhibit glycogen breakdown. This causes excess glycogen to accumulate in muscles, liver, kidney, and other organ tissue — which is toxic.
In most cases, glycogen storage disease starts early in life. Depending on the type of GSD, the symptoms range from mild to severe — with muscle pain, enlarged organs, stunted growth, low blood sugar, and muscle weakness being a few of the most common side effects[*].
How Your Body Stores Glucose
When you eat a meal — especially a carb-rich meal — much of that meal ends up in your bloodstream as glucose, a simple sugar that provides your cells with energy[*].
This rise in blood glucose, or blood sugar, then signals your pancreas to release insulin — your blood sugar regulation hormone.
Insulin’s job is critical — it ushers glucose out of your blood and into your cells for energy or stores it away for later. This not only provides you with the fuel you need to function, it also maintains healthy blood sugar levels.
Since high blood sugar, or hyperglycemia, is linked to most chronic diseases and metabolic disorders — you definitely want balanced blood sugar and insulin-sensitive cells[*][*][*].
After insulin sends its message to get glucose out of your blood, your cells store glucose in one of two forms: glycogen or fat. When insulin is working correctly — when you’re insulin sensitive — most of your excess blood glucose gets stored as the first form (glycogen) in muscle and liver cells.
Glycogen storage requires certain enzymes, including:
- Glycogen synthase
- Glycogen debranching enzyme
- Acid-alpha-glucosidase
Healthy glycogen storage enzymes promote healthy amounts of glycogen in your cells.
How Glycogen Storage Works
Here’s an oversimplification of glycogen storage from start to finish[*]:
- Your blood sugar rises after eating
- Insulin commands your cells to store any excess glucose as glycogen
- Glucose gets converted to glucose-6-phosphate
- Glucose-6-phosphate gets converted to glucose-1-phosphate
- Glucose-1-phosphate gets converted to UDP-glucose
- UDP-glucose becomes glycogen thanks to the enzyme glycogen synthase
Once stored, glycogen sits in your muscle and liver cells until you have low blood sugar — during a fast or intense exercise session, for instance.
Then, to raise your low blood sugar, glycogen converts into glucose so your body can use it for energy — a process called glycogenolysis.
How Glycogenolysis Works
Here’s what glycogenolysis looks like under normal conditions[*]:
- Your blood sugar drops because you haven’t eaten for a while, are intentionally fasting, or an intense workout
- Low blood sugar signals glycogen stores to convert into glucose-1-phosphate
- Glucose-1-phosphate converts into glucose-6-phosphate
- Your body uses glucose-6-phosphate for energy (glycolysis) or turns it into glucose and releases it into the blood via an enzyme called glucose-6-phosphatase
At least that’s how glycogen storage and breakdown normally work. But when someone has glycogen storage disease, one or more of these steps is disrupted — and this leads to problems with muscle, liver, heart, and other organ tissue.
Read Next: Glucose Ketone Index (GKI): What It Is and Quick Calculator
Types of Glycogen Storage Disease
Believe it or not, there are at least 16 types of GSD (type 0 through 15). This article, however, will only cover the first eight, not counting the subtypes within each type.
Recall that glycogen storage disease results from genetic mutations that disrupt the storage and breakdown of glycogen. Types of GSD, then, are differentiated by specific genetic mutations.
Type 0 (GSD 0)

Type I (GSD1, Von Gierke Disease)

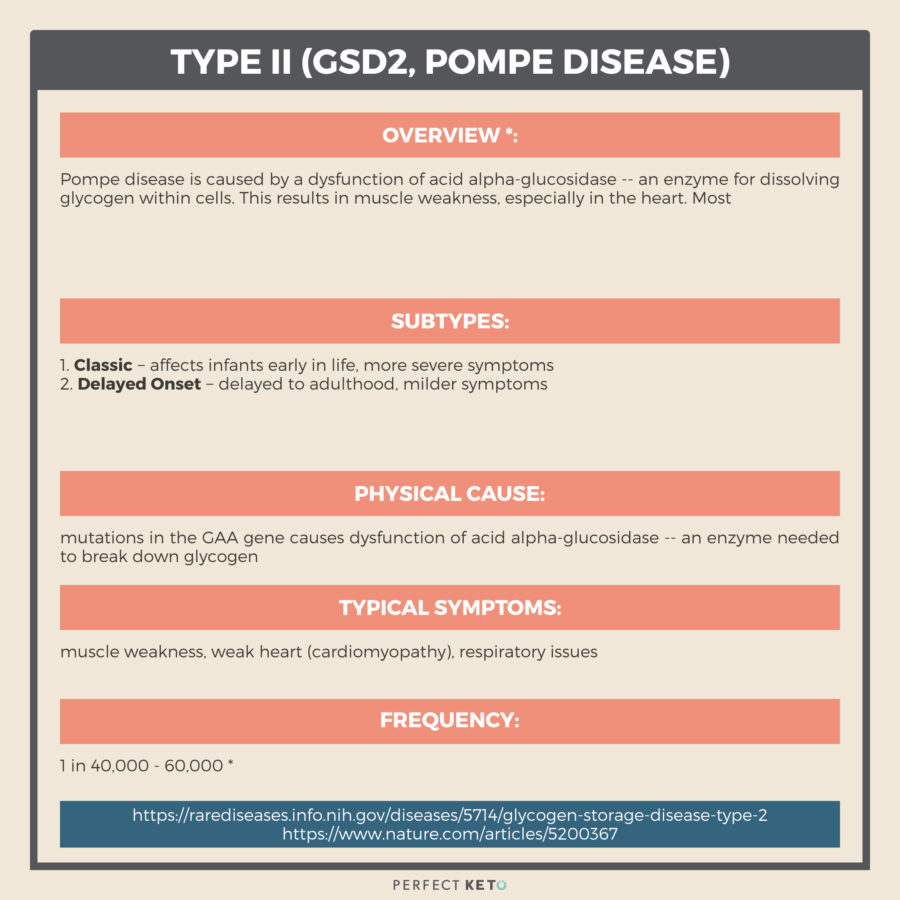
Type II (GSD2, Pompe Disease)
Type III (GSD3, Cori Disease)

Type IV (Andersen disease, GSD IV)

Type V (GSDV, McArdle Disease)

Type VI (GS DVI, Hers Disease)

Type VII (GSDVII)

How Common Is Glycogen Storage Disease?
There are at least seven other types of GSD described in the literature. And these types have similar roots to types I through VII.
The truth is: GSDs of all types are probably underdiagnosed. As genetic testing becomes more common, and as research progresses, the number of reported cases may increase.
And with more diagnoses, it’s also likely that researchers will discover more (and better) therapies.
But GSD isn’t necessarily a hopeless prognosis. There are promising therapies — both pharmaceutical and nutritional — for this group of genetic disorders. And they deserve a mention here.
Drugs And Gene Therapy For GSD
GSD treatment usually revolves around symptom-management, but scientists have recently developed therapies that go beyond managing symptomatology. Below are a few of these therapies.
But first, a standard disclaimer. This article does not take the place of medical advice. Please consult a healthcare professional for that.
#1 Lumizyme
In 2014, the FDA approved the drug Lumizyme to treat Pompe disease (GSD2). Lumizyme is an is enzyme replacement therapy that replaces the glycogen breakdown enzyme that people with Pompe disease lack[*].
Also known as alglucosidase alfa, Lumizyme shows promise for treating both Pompe disease and Cori disease in clinical trials[*][*].
#2 Rapamycin
Rapamycin is a drug that regulates the mammalian target of rapamycin (mTor) pathway. In brief, the mTor pathway governs cell metabolism, growth, and much more. Relevant here: the mTor pathway also affects the enzyme glycogen synthase, responsible for proper glycogen storage
Skipping to the punchline: rapamycin given to dogs with Cori disease (GSD IIIa) inhibited glycogen synthase, reduced muscle and liver glycogen, and prevented damage to both liver and muscle tissue[*].
# 3 Gene Therapy
Want to modify gene expression in your average mammal? It’s possible. Just inject that mammal with a virus.
You read that right. Injecting dogs, cats, mice, and even sheep with adeno-associated virus (AAV) has delivered promising results for multiple forms of GSD, including von Gierke disease, Pompe disease, and McArdle disease[*].
What about human trials? Well, five people with Pompe disease showed modest improvement, with few side effects, from AAV therapy[*].
Yet the most accessible therapy for GSD isn’t a drug or a virus. It’s nutrition.
Sugar And GSD
If you or a loved one suffers from glycogen storage disease, the first thing you can do is address sugar intake.
Sugary, highly processed, and high-carb foods will raise your blood sugar, aka blood glucose. High blood glucose is a huge problem if your body has a hard time breaking down, storing, or releasing glycogen.
Yes, people with GSD will store excess glucose as glycogen; but, unfortunately, that glycogen can’t convert back to glucose for energy later on. Which means glycogen builds to toxic levels.
Less glycogen is usually a good thing for people with GSD, so it’s logical that minimizing the blood sugar response (called hypoglycemia treatment) can help[*]. With less sugar in the blood, less glycogen gets stored.
The obvious way to reduce sugar in the blood? Eat fewer carbs.
And so for treating GSD, researchers are focusing on very low-carb, high-fat diets to keep glycogen storage at a minimum.
The Keto Diet For GSD
Your body tends to run on glucose. But in a low-sugar, hypoglycemic state, your body doesn’t just call it quits. Instead, it starts producing ketones from dietary and stored fat — your backup energy source.
Ketones, like glucose, are eventually converted to ATP (aka cellular energy) through a process called the Krebs cycle. But unlike glucose, ketones don’t spike your blood sugar, and don’t need to be stored as glycogen.
Because of this, the ketogenic diet — a high-fat, low-carb nutritional plan that gets you burning fat over sugar — has shown promise for treating glycogen storage disease. Here are some examples:
- A high-fat diet lessened myopathy (muscle weakness) in two boys with Cori disease (type III GSD) over the course of about 2.5 years. This benefit stopped when the high-fat diet stopped and resumed when they resumed the high-fat diet[*].
- A high-fat diet alleviated cardiomyopathy (heart muscle weakness) in two siblings with Cori disease over the course of one year. Cardiac enzymes, along with signs of congestive heart failure, both improved measurably[*].
- A ketogenic diet was used to treat a patient with GSD V, or McArdle disease — resulting in less muscle pain and weakness[*]
Admittedly, these trials are small, and more research is required — but a ketogenic or very low-carb diet may offer some hope to families dealing with GSD.
Glycogen Storage Disease Next Steps
If you think your child or a loved one suffers from glycogen storage disease, the first thing to do is consult your doctor. Depending on the type of GSD, there are one or more potential drug therapies on the market or in the pipeline.
You might also research nutritional options for GSD. Research on low carb keto diets is still early, but several small case studies have shown promise.
To learn more about keto, you can read our free startup guide here.
READ NEXT: Ketosis For Children